 |
 |
 |
 |
 |
|
The idea of the method is that a material is represented as a set of interacting particles. The concept is close to the Molecular Dynamics (MD) method, where the material is modelled by a large number of atoms (molecules) interacting with each other via prescribed nonlinear force laws; trajectories of each particle are followed through time by integrating the Newton's classical equations of motion. The interaction forces usually contain potential and dissipative components. One of the simplest examples of interparticle potential is the Lennard-Jones potential. The main feature of this potential is that it produces attraction at long range and repulsion at short range.
The main difference of my method from the conventional MD is that particles are considered not as atoms or molecules but as elements of mesoscale level, such as material grains. For the material simulation empirical interparticle potentials can be chosen to give the best agreement with the elastic and strength constants.
Under an action of the interaction forces the particles can coalesce forming material grains. Below you can see such grains cobained from small number of particles.
 |
 |
 |
 |
 |
Some larger grains. You can press any picture to enlarge it.
 |
 |
 |
 |
 |
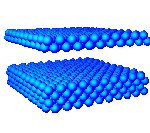
The particles can form crystal lattices. An example of a crystal with FCC (face centered close packed) lattice is depicted below. The particle radii are shown as if they were two times smaller then in the above pictures, so that the inner structure of the crystal is better visible.

For more information look at the next page, please!
Designed by Julia Asaturova
